Procesamiento de datos 16S
Seleccione la opción procesar, y se despliega “choose command” este le dará una lista de pasos de procesamientos disponibles.
Para los objetos 16S “FASTQ”, el único comando disponible es “Split libraries FASTQ”. Convierte los datos sin procesar de FASTQ en el formato de archivo utilizado por Qiita para un análisis más detallado (puede leer más sobre este tipo de archivo aquí ).
Seleccione el paso "Split libraries FASTQ". Ahora, podrá seleccionar la combinación específica de parámetros que se utilizará para este paso en el menú desplegable "Parameter set".

Para los archivos de práctica se eligio “Multiplexed FASTQ; Golay 12 base pair reverse complement mapping file barcodes with reverse complement barcodes” más información (aquí), seguido clic en “Add command” seguido de clic en “run” y esperamos que termine de realizar el primer análisis en la red, al seleccionar la figura del "Split libraries FASTQ" nos muestra el estado del análisis.
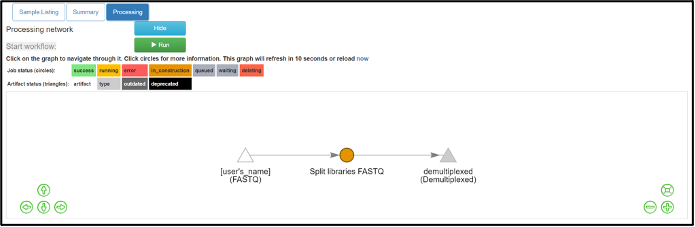
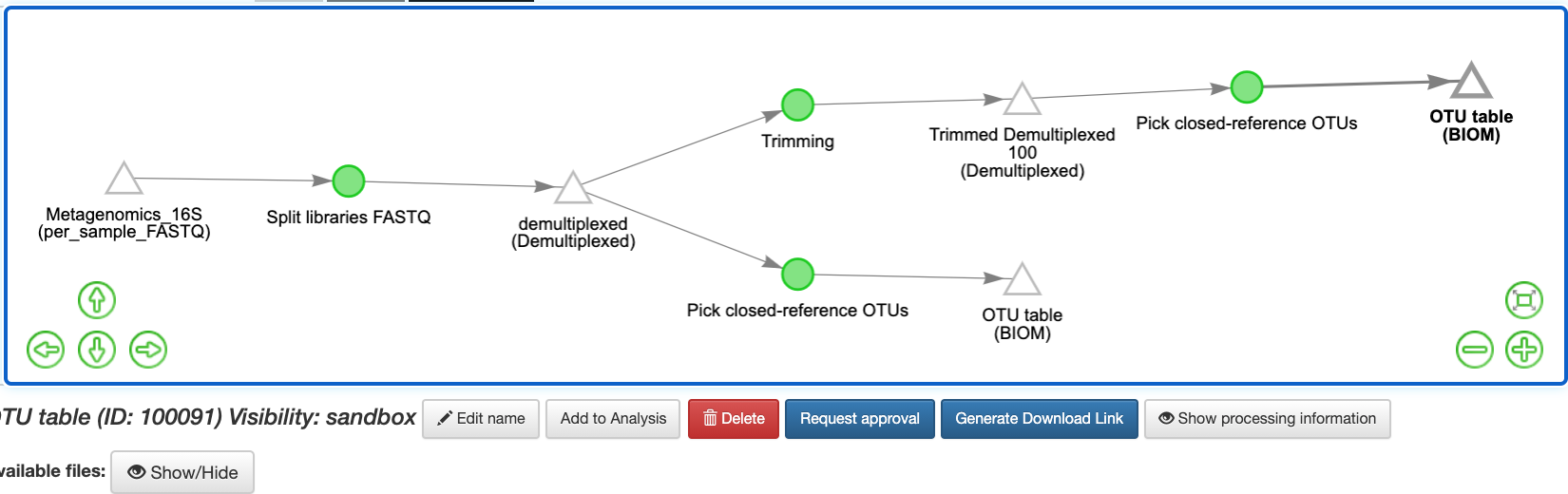
Una vez concluído el análisis se pondrá en verde la red y mostrará los resultados:
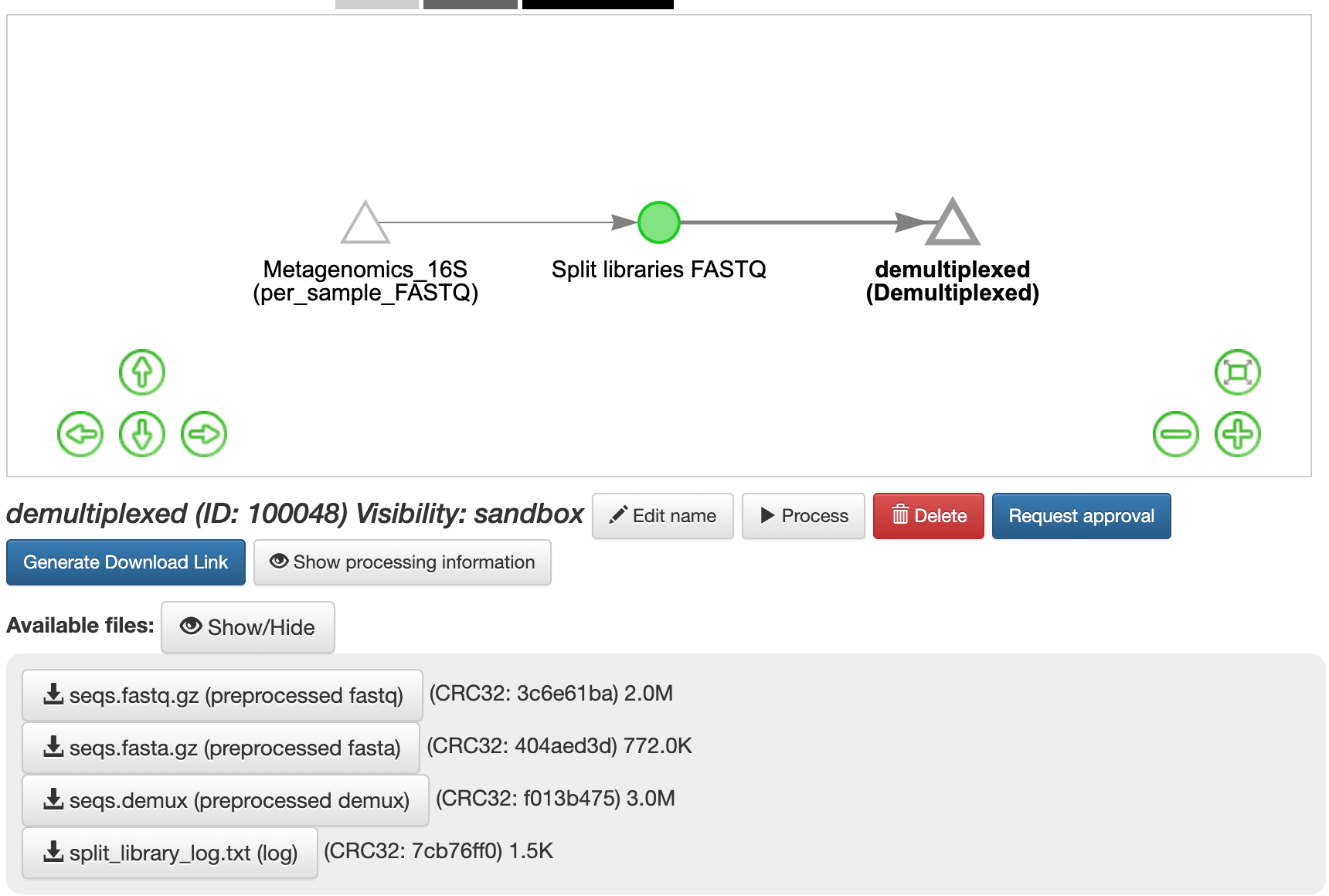

A continuación, queremos recortar a una longitud particular, para asegurarnos de que nuestras muestras sean comparables a otras muestras que ya están en la base de datos. Vuelva a hacer clic en "demultiplexed(demultiplexed)". Esta vez, seleccione la operación de “Trimming”. Actualmente, hay siete opciones de longitud de recorte. Nosotros elegimos "100 pares de bases", que recorta a los primeros 100 pb, para esta ejecución, y haga clic en "Add Command" y “Run”.
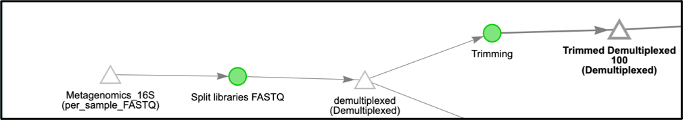
Para eliminar las secuencias que no coinciden con las que se encuentran en las base de datos se selecciona el objeto “Trimmed Demultiplexed (Demultiplexed)” y selecciona en el área de comando la opción “Pick Closed-Reference OTUs”, utilizando los parametros por defecto que son relativamente pequeños. Qiita utiliza de manera predeterminada la base de datos GreenGenes. Una vez listo se agrega el comando y le damos clic a “Run”.
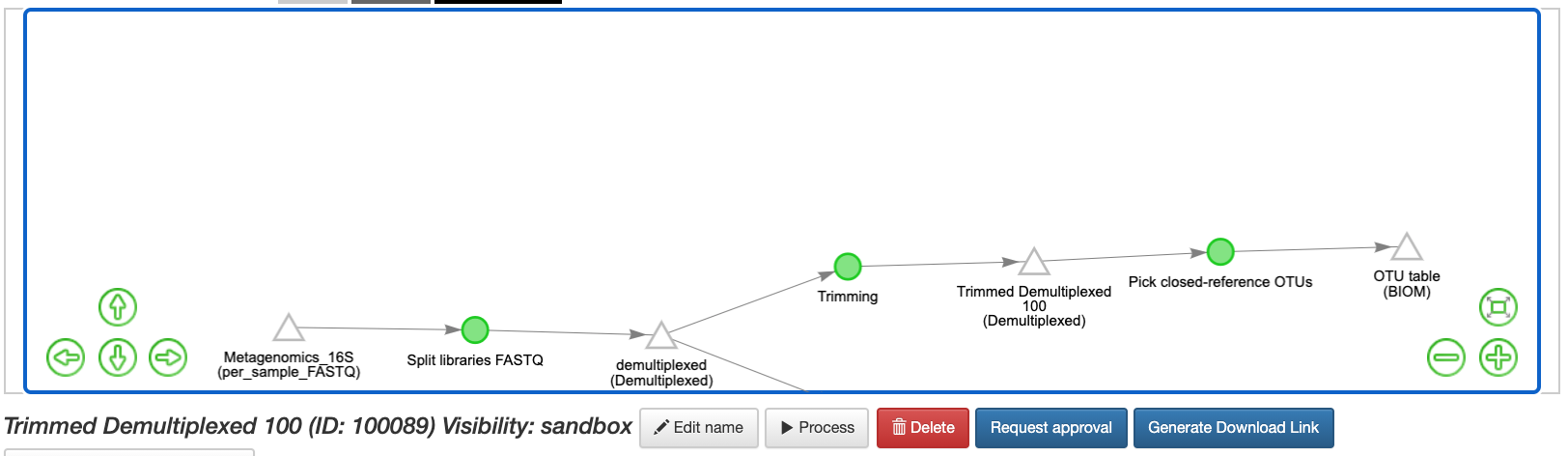
Una vez que se han generado los objetos, puede generar resúmenes para ellos tal como lo hizo para el objeto "FASTQ" original.
El resumen del objeto "demultiplexed (demultiplexed)" le brinda información sobre la longitud de las secuencias en el objeto:
Una vez creado el objeto después del múltiplexado, podemos seleccionar este objeto y dar clic “Add to Analysis” y esto generá un nuevo análisis, que aparecerá en la barra superior derecha.
Hacer clic en el icono lo llevará a una página donde puede refinar las muestras que desea incluir en su análisis.

Opcionalmente, puede excluir muestras particulares de este conjunto haciendo clic en "Mostrar / Ocultar muestras", que mostrará el nombre de cada muestra individual junto con la opción "eliminar". (Quitarlos aquí los ocultará del análisis, pero no afectará a los archivos subyacentes de ninguna manera).
Haga clic en el botón " Create Analysis", introduzca un nombre y una descripción, luego haga clic en " Create Analysis".

Esto lo lleva a la página de la red de procesamiento. Esto puede tardar de 2 a 5 minutos en cargarse. Cuando se haya cargado, puede analizar los datos.
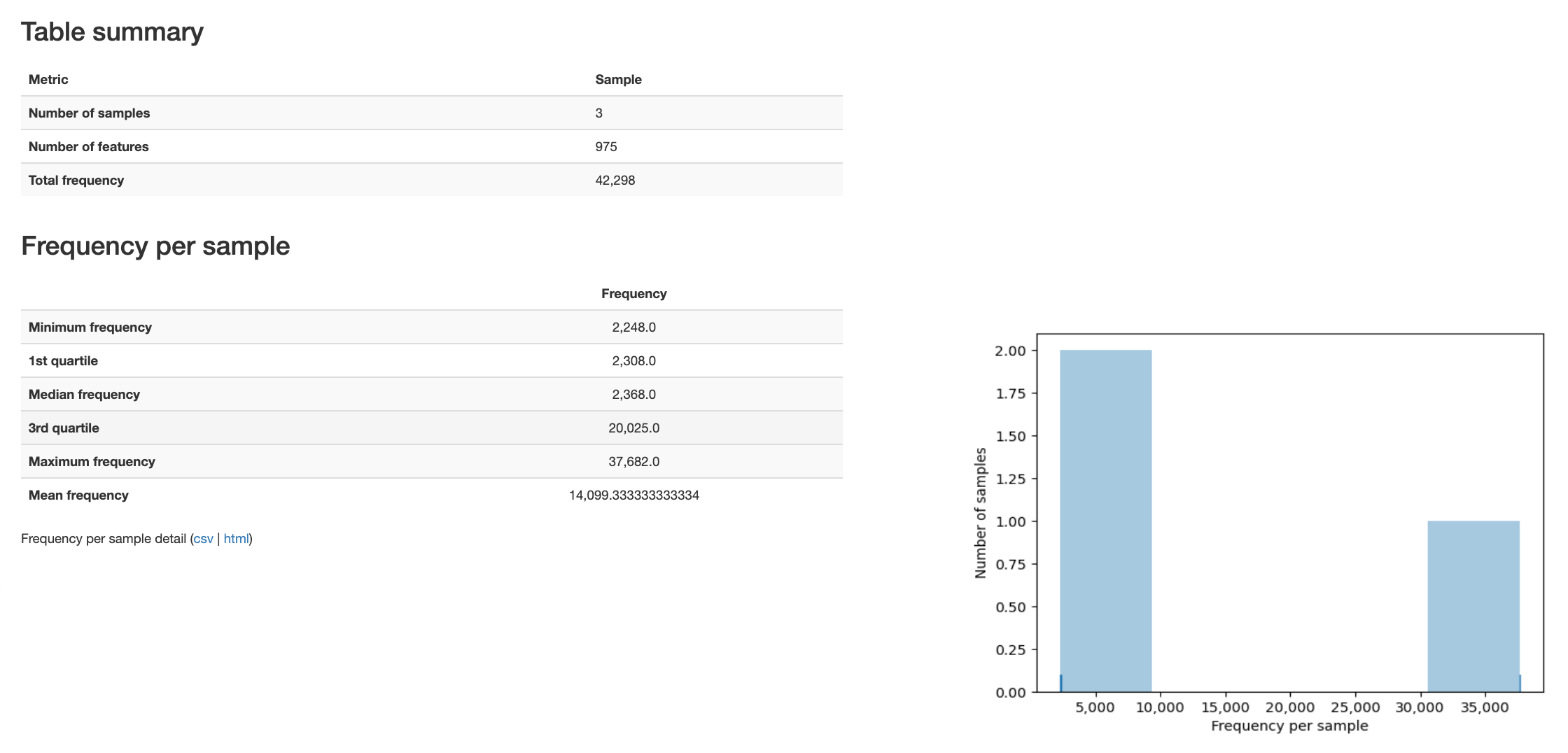
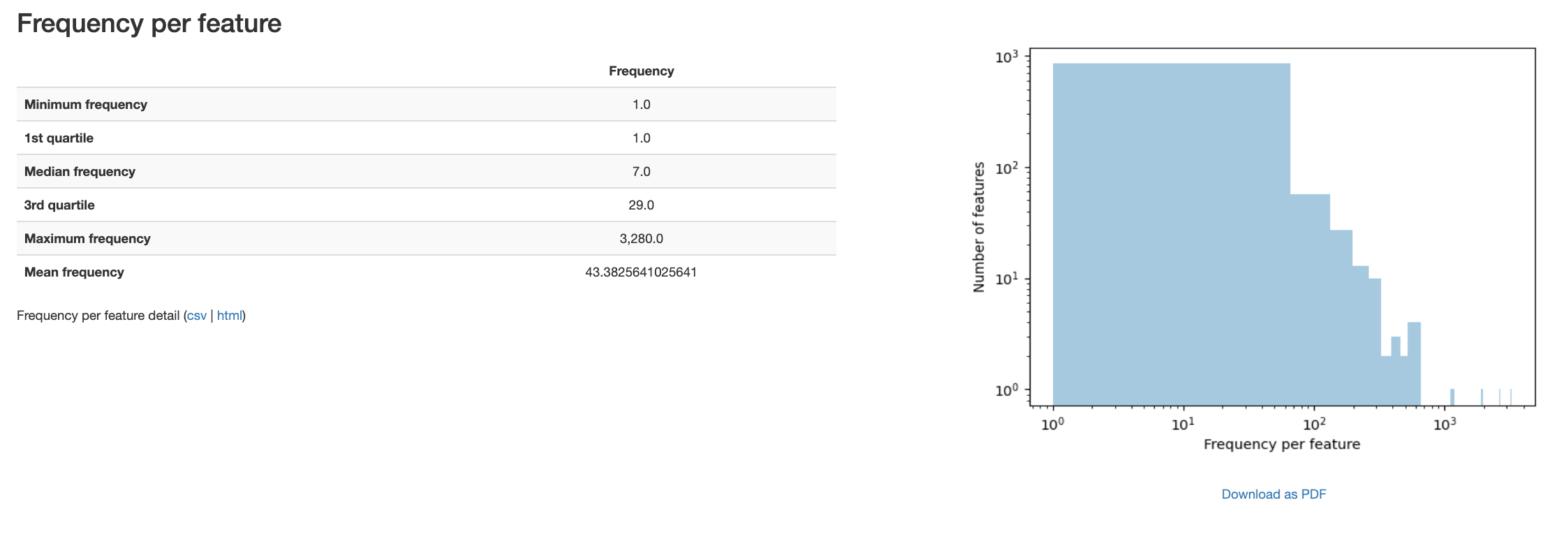
Antes de procesar los datos, veamos un vistazo al resumen del contenido del archivo biom. Seleccione el artefacto "dflt_name (BIOM)" para ver un resumen de este archivo que muestra un resumen de la tabla, detalles sobre la frecuencia por muestra, histograma del número de características por muestra:
Como pueden ver, este archivo contiene 3 muestras con aproximadamente 42298 características.
Ahora comienza análizar estas muestras. Se selecciona "dflt_name (BIOM)" y luego seleccione "Process". Esto nos llevará a la página de selección de comandos. Una vez allí, se puede acceder a la pestaña desplegable de comandos que mostrará treinta y seis acciones.

El texto entre paréntesis son los comandos reales de QIIME2. Ahora se utilizará algunos de los comandos más utilizados que le permitirán generar resúmenes, vrificar datos y calcular estadísticas para aprovechar al máximo los datos.