Ensamblaje
"Co-ensamblaje" es la realización de un ensamblaje donde los archivos de entrada se leerían de múltiples muestras. Esto está en contraste con hacer un ensamblaje independiente para cada muestra, donde la entrada para cada uno sería solo la lectura de esa muestra individual.
Los tres ventajas principales incluyen:
1) Mayor profundidad de lectura, esto permite tener un ensamblaje más robusto que capture más la diversidad.
2) Facilita la comparación entre muestras al darle un conjunto de referencia.
3) Mejora sustancialmente su capacidad de recuperar genomas dentro de los metagenomas gracias a la cobertura diferencial.
Hay muchos ensambladores, y ninguno es malo cada uno tiene sus ventajas dependiendo al conjunto de datos que se quiera analizar, es decir; Algunos ensambladores funcionan mejor para algunos conjuntos de datos, y otros funcionan mejor para otros. Esto no quiere decir que todos sean mágicamente igual de buenos en todos los sentidos, pero la mayoría de los que lo siguen superarán a todos los demás bajo ciertas condiciones. Los ensambladores más utilizados son SPAdes , Megahit, Minia , y para verificar los datos con QUAST (para ensamblaje de genoma individual) o MetaQUAST (para ensamblajes de metagenoma). Elegir el "mejor" ensamblaje no es sencillo y puede depender de lo que esté haciendo.
Co-ensamblaje
"Co-ensamblaje" es la realización de un ensamblaje donde los archivos de entrada se leerían de múltiples muestras. Esto está en contraste con hacer un ensamblaje independiente para cada muestra, donde la entrada para cada uno sería solo la lectura de esa muestra individual.
Los tres ventajas principales incluyen:
1) Mayor profundidad de lectura, esto permite tener un ensamblaje más robusto que capture más la diversidad.
2) Facilita la comparación entre muestras al darle un conjunto de referencia.
3) Mejora sustancialmente su capacidad de recuperar genomas dentro de los metagenomas gracias a la cobertura diferencial.
Hay muchos ensambladores, y ninguno es malo cada uno tiene sus ventajas dependiendo al conjunto de datos que se quiera analizar, es decir; Algunos ensambladores funcionan mejor para algunos conjuntos de datos, y otros funcionan mejor para otros. Esto no quiere decir que todos sean mágicamente igual de buenos en todos los sentidos, pero la mayoría de los que lo siguen superarán a todos los demás bajo ciertas condiciones. Los ensambladores más utilizados son SPAdes , Megahit, Minia , y para verificar los datos con QUAST (para ensamblaje de genoma individual) o MetaQUAST (para ensamblajes de metagenoma). Elegir el "mejor" ensamblaje no es sencillo y puede depender de lo que esté haciendo.
METASPADES
| metaspades.py -o assembly_metaspades --pe1-1 bbduck-mix_1.fastq --pe1-2 bbduck-mix_2.fastq |

En este caso, así es como se ejecutaría el comando con el ensamblador metaSpades. Pero incluso con el conjunto de datos pequeño que estamos usando, esto lleva tiempo en ejecutarse.
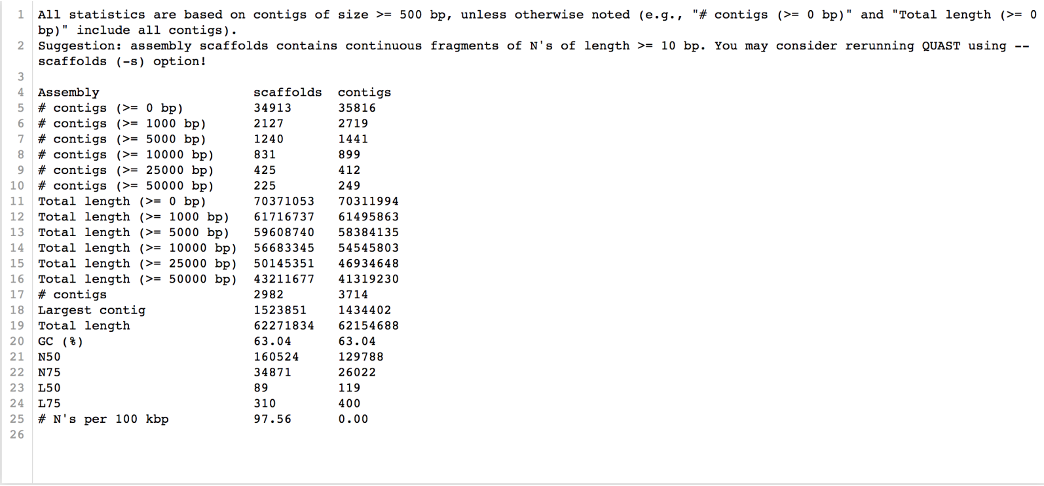
QUAST
| quast.py assembly_metaspades/scaffolds.fasta assembly_metaspades/contigs.fasta -o quast-27-19 |
REFORMATEAR
Si abrimos el archivo contigs.fa se puede ver que hay espacios y algunos otros caracteres especiales en los encabezados (las líneas que comienzan con ">"). Esto puede ser problemático para algunas herramientas, y es mejor tener encabezados simplificados si se puede. Hay muchas maneras de hacer modificaciones como esa, y algunos scripts ya existen. Anvi'o tiene uno, que realiza la simplificación de los nombres de contigs en él archivo y elimina algunos de los contigs cortos, este comando lo utilizaremos. También vamos a filtrar los contigs que sean más cortos que 2500 bps. Si desea hacer esto normalmente o no, depende del investigador, sin embargo siempre es recomendable usar entre 2500 y 5000 bps como mínimo a la hora de emsamblar los MAGS, ya que tienen un mayor sentido biológico.
| anvi-script-reformat-fasta contigs.fasta -o reformat_contigs_fasta/contigs.fa --min-len 2500 --simplify-names --report name_conversions.txt |
Desglose del código:
▪ anvi-script-reformat-fasta es el comando, especifica el programa que estamos usando.
▪ El primer argumento "posicional" (no se necesita marca) es especificar el archivo de entrada (nuestro ensamblado).
▪ -o especifica el nombre del archivo de salida.
▪ -l especifica que queríamos filtrar secuencias con menos de 2500 bps.
▪ --simplify-names le dice al programa que queremos que le dé a todas las secuencias un encabezado "limpio" (sin caracteres especiales).
MAPEANDO LAS LECTURAS DEL ENSAMBLAJE
El mapear las lecturas para el co-ensamblaje de nuestro ensamblaje nos da información de "cobertura" para cada contig en cada muestra, que como se discutió anteriormente nos ayudará con nuestros esfuerzos para recuperar los MAGs y saber en qué muestra se encuentra cada contig ensamblado.
En está sección se utiliza la herramienta bowtie2 para hacer el mapeo, y como primer paso se necesita crear un índice del co-ensamblaje:
| bowtie2-build reformat_contigs_fasta/contigs.fa mapping/contigs |
Y aquí es donde se mapea las lecturas de las muestras individuales a un ensamblaje en conjunto, mapear y convertir los formatos de archivo a lo que necesitamos tomaría unos minutos para ejecutar todas las muestras.
El formato Sam es un archivo que se utiliza para almacenar grandes alineamientos de secuencias de nucleótidos, compatible con muchas herramientas de alineación de secuencias, casi no utiliza memoria y permite que el archivo se indexe por posición genómica para recuperar las lecturas alineadas.
| Bowtie2 –threads 10 -x assembly_metaspades/mapping/contigs -1 bbduck-mix_1.fastq -2 bbduck-mix_2.fastq -S assembly_metaspades/mapping/samples.sam |

Desglose del código:
bowtie2 es el comando, especifica el programa que estamos usando.
● -x Se especifica el "nombre base" del índice que acabamos de construir, que es "ensamblado.
● -1 Especifica las lecturas directas de la muestra.
● -2 Especifica las lecturas inversas.
● -p Especifica cuántos cpus se van a utilizar.
● -S Especifica el nombre del archivo “sam” de salida que queremos crear ( Sequence Alignment Map).
Convertir archivo Sam a archivo Bam (Binary Alignment Map)
| samtools view -F 4 -b S samples.sam > samples-RAW.bam |
Desglose del código:
samtools es el programa principal, especifica el programa que estamos usando.
● View es el subprograma de lo samtools que estamos llamando.
● -b le dice al programa que queremos que la salida esté en formato bam.
● -o especifica el nombre del archivo de salida.
● el último argumento "posicional" (no se necesita marca) le dice el archivo de entrada, que es nuestro archivo sam para esta muestra.
Este comando genera un archivo .bam y sus índices correspondientes llamados .bai en el directorio de trabajo.